Marfan Syndrome is a hereditary connective tissue disorder caused primarily by mutations in the FBN1 gene. It affects approximately 1 in 5,000 individuals worldwide and is associated with life-threatening cardiovascular complications, particularly thoracic aortic aneurysm and dissection.
While historically described as a structural connective tissue disease, Marfan Syndrome is now recognised as a complex systemic disorder driven by dysregulated extracellular matrix (ECM) biology, transforming growth factor-β (TGF-β) signalling and oxidative stress pathways.
Recent advances in proteomics and antibody microarray technologies are enabling molecular risk stratification and paving the way for Precision Medicine in Marfan Syndrome.
I. Marfan Syndrome: Genetic Causes and Pathophysiology
Marfan Syndrome is an autosomal dominant disorder caused in most cases by mutations in the FBN1 gene on chromosome 15. The gene encodes fibrillin-1, a structural glycoprotein that is essential for the integrity and elasticity of connective tissue.
Beyond its mechanical role, fibrillin-1 regulates the bioavailability of TGF-β, a key signalling molecule controlling cell proliferation, extracellular matrix remodelling and inflammation.
When fibrillin-1 is deficient or dysfunctional, excessive TGF-β signalling occurs. This molecular imbalance contributes directly to vascular wall degeneration, aneurysm formation and multisystem manifestations.
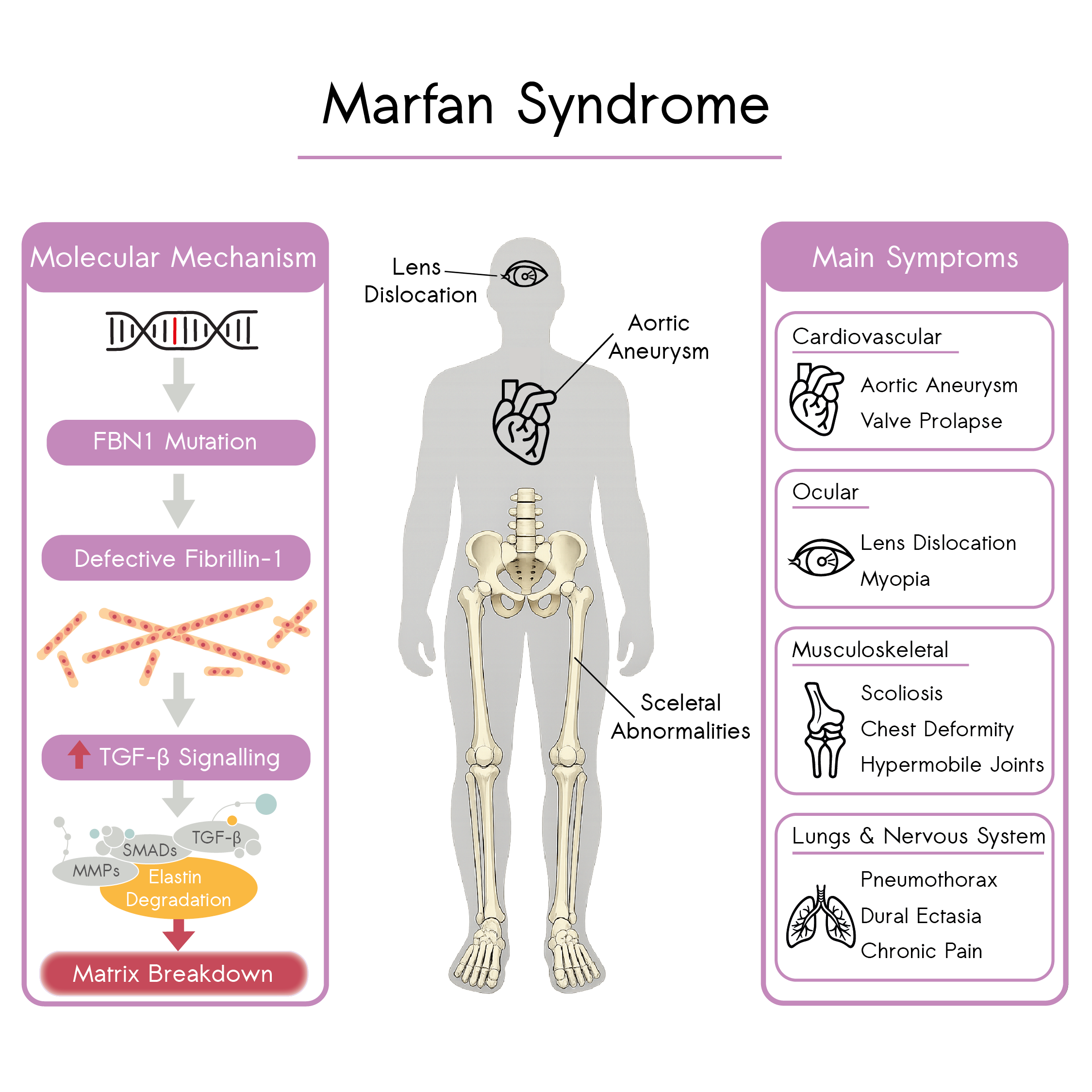
Molecular mechanisms and systemic manifestations of Marfan Syndrome: FBN1 mutations lead to defective fibrillin-1, dysregulated TGF-β signalling and extracellular matrix breakdown, driving aortic aneurysm formation and multisystem involvement affecting the cardiovascular, ocular, musculoskeletal and pulmonary system.
II. Molecular Mechanisms: TGF-β, MMPs and Vascular Degeneration
The molecular pathogenesis of Marfan Syndrome extends far beyond structural instability of connective tissue.
II.1 Dysregulated TGF-β Signalling
Fibrillin-1 deficiency leads to increased levels of active TGF-β. In vascular smooth muscle cells (VSMCs), this results in:
- Enhanced phosphorylation of Smad2/3 (canonical pathway activation)
- Altered extracellular matrix gene expression
- Increased inflammatory and oxidative signalling
II.2 Matrix Metalloproteinase Activation
Upregulation of matrix metalloproteinases, particularly MMP-2 and MMP-9, accelerates elastin degradation within the aortic wall. This degradation weakens elastic lamellae and promotes progressive thoracic aortic aneurysm formation.
II.3 Non-Canonical Signalling Pathways
Proteomic studies in Marfan mouse models have identified TGF-β-dependent activation of the integrin-β3–mTORC2/Rictor axis in the ageing aorta. This non-canonical signalling pathway modulates VSMC proliferation and migration and contributes to aneurysm progression.
Together, these mechanisms explain why Marfan Syndrome is now regarded as a systemic signalling disorder rather than a purely structural disease.
III. Clinical Symptoms of Marfan Syndrome: Cardiovascular, Ocular and Skeletal Features
Marfan Syndrome comes with considerable phenotypic variability, even among individuals carrying FBN1 mutations. Classic features include:
| Clinical Symptoms | Phenotypic Symptoms |
| Aortic root dilation | Tall stature with slender body |
| Aortic aneurysm | Long arms and legs (arm span > height) |
| Aortic dissection | Arachnodactyly (“spider fingers”) |
| Mitral valve prolapse | Hyperextensible joints (joint hypermobility) |
| Lens dislocation (Ectopia lentis) | Pectus excavatum (funnel chest) |
| Severe myopia | Pectus carinatum (pigeon chest) |
| Spontaneous pneumothorax (lung collapse) | Scoliosis (spinal curvature) |
| Dural ectasia (expansion of dural sac) | Stretch marks (striae) without cause |
IV. Current Disease Diagnosis: Ghent Criteria and Genetic Testing
Diagnosis is currently based on the revised Ghent nosology (2010), integrating:
- Family history
- Aortic root measurements
- Systemic clinical features
- Identification of a pathogenic FBN1 mutation
However, genotype alone does not reliably predict disease severity or aortic progression rate. Individuals with identical mutations may exhibit markedly different outcomes. This limitation underscores the importance of molecular profiling approaches.
V. Marfan Syndrome Biomarkers: Proteomics for Risk Stratification
One of the central challenges in Marfan Syndrome management is predicting aortic aneurysm progression and dissection risk. Proteomics research has identified several promising circulating biomarkers.
V.1 TGF-β as a Circulating Marker
Elevated plasma TGF-β levels correlate with aortic root dilatation and faster aortic growth rates. TGF-β reflects underlying molecular dysregulation, although it is not yet sufficient as a stand-alone predictor of therapeutic response.
V.2 MMP-2 and MMP-9 in Aortic Remodelling
Increased levels of MMP-2 and MMP-9 in tissue and plasma correlate with elastin fragmentation and aneurysm expansion. These proteases represent both biomarkers of progression and potential therapeutic targets.
V.3 3-Nitrotyrosine: A Marker of Oxidative Stress in Marfan Syndrome
3-nitrotyrosine (3-NT) is a stable oxidative protein modification formed through peroxynitrite-mediated nitration. Elevated 3-NT levels indicate chronic oxidative stress, endothelial dysfunction and vascular degeneration.
In thoracic aortic aneurysm tissue and plasma, increased 3-NT correlates with disease progression. Importantly, oxidative modifications may occur before detectable structural changes, making 3-NT a potential early biomarker of vascular damage.
Multiplex antibody microarrays incorporating anti-3-nitrotyrosine detection allow simultaneous analysis of oxidative stress markers alongside TGF-β- and MMP-associated pathways. This systems-level profiling approach strengthens biomarker discovery and validation.
V.4 Proteoglycans and ECM Markers
Lumican and decorin, small leucine-rich proteoglycans involved in collagen organisation and TGF-β modulation, show altered plasma levels in acute aortic dissection and extracellular matrix remodelling. Their concentrations may reflect aortic stiffness and impending vascular instability.
V.5 Emerging Molecular Biomarkers
Additional candidates include deregulated microRNAs such as miR-29b and miR-145, which are associated with extracellular matrix remodelling and thoracic aortic aneurysm progression. Circulating elastin fragments reflect elastic lamella breakdown and correlate with aneurysm growth and dissection risk.
VI. Antibody Microarrays and High-Throughput Proteomics in Marfan Research
High-throughput antibody microarrays enable the parallel analysis of thousands of proteins from minimal sample volumes. Compared with traditional single-analyte assays, multiplex proteomics provides a comprehensive view of cytokines, extracellular matrix components, inflammatory mediators and oxidative stress markers as 3-NT in a single experiment.
This approach facilitates:
- Identification of molecular signatures
- Early risk stratification
- Therapy response monitoring
- Patient subgroup classification
Such technologies are central to implementing Precision Medicine strategies in Marfan Syndrome.
VII. Current Treatment of Marfan Syndrome
Modern management has significantly improved life expectancy. Standard treatment aims to reduce haemodynamic stress on the aorta and slow aneurysm progression.
Therapeutic strategies include beta-blockers such as atenolol, angiotensin receptor blockers such as losartan to attenuate TGF-β signalling, regular imaging surveillance and prophylactic surgical repair when indicated.
Despite advances in pharmacological management, no curative therapy currently exists.
VIII. Molecular Stratification and Emerging Therapeutic Strategies
The future of Marfan Syndrome care lies in molecularly informed, individualised treatment strategies.
Genetics-based stratification may differentiate mutation types associated with more severe phenotypes, informing monitoring frequency and treatment decisions.
VIII.1 Microarrays as the Next Step Toward Precision Monitoring
Integration of untargeted mass spectrometry, targeted antibody microarrays and detailed FBN1 mutation profiling may enable identification of patient subgroups with distinct aneurysm progression trajectories. Such stratification could guide selection of beta-blockers, angiotensin receptor blockers, TGF-β-modulating agents or emerging targeted therapies.
Experimental CRISPR-based base editing approaches have demonstrated correction of FBN1 point mutations in vitro and represent a potential long-term causal strategy. Patient-specific induced pluripotent stem cell models further allow personalised vascular smooth muscle cell studies and drug response testing, advancing the implementation of Precision Medicine.
IX. Towards Precision Medicine in Marfan Syndrome
Marfan Syndrome is no longer viewed solely as a monogenic connective tissue disorder. It is increasingly defined by its molecular signatures — signalling dysregulation, ECM remodelling and oxidative stress.
The integration of genomic insights with high-throughput proteomics enables a shift from reactive management towards proactive, risk-adapted care. In this evolving framework, Precision Medicine is not an abstract concept, but a tangible clinical objective